Flory–Huggins solution theory
Flory-Huggins solution theory is a mathematical model of the thermodynamics of polymer solutions which takes account of the great dissimilarity in molecular sizes in adapting the usual expression for the entropy of mixing. The result is an equation for the Gibbs free energy change 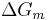 for mixing a polymer with a solvent. Although it makes simplifying assumptions, it generates useful results for interpreting experiments. The thermodynamic equation for the Gibbs free energy change accompanying mixing at constant temperature and (external) pressure is
for mixing a polymer with a solvent. Although it makes simplifying assumptions, it generates useful results for interpreting experiments. The thermodynamic equation for the Gibbs free energy change accompanying mixing at constant temperature and (external) pressure is
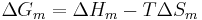
A change, denoted by  , is the value of a variable for a solution or mixture minus the values for the pure components considered separately. The objective is to find explicit formulas for
, is the value of a variable for a solution or mixture minus the values for the pure components considered separately. The objective is to find explicit formulas for 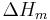 and
and 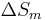 , the enthalpy and entropy increments associated with the mixing process.
, the enthalpy and entropy increments associated with the mixing process.
The result obtained by Flory[1] and Huggins[2] is
![\Delta G_m = RT[\,n_1\ln\phi_1 %2B n_2\ln\phi_2 %2B n_1\phi_2\chi_{12}\,] \,](/2012-wikipedia_en_all_nopic_01_2012/I/aea39e7f9ec0ac97923925da507d0a8f.png)
The right-hand side is a function of the number of moles 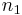 and volume fraction
and volume fraction 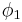 of solvent (component
of solvent (component 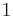 ), the number of moles
), the number of moles 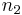 and volume fraction
and volume fraction 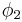 of polymer (component
of polymer (component 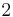 ), with the introduction of a parameter chi
), with the introduction of a parameter chi 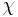 to take account of the energy of interdispersing polymer and solvent molecules.
to take account of the energy of interdispersing polymer and solvent molecules. 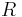 is the gas constant and
is the gas constant and 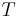 is the absolute temperature. The volume fraction is analogous to the mole fraction, but is weighted to take account of the relative sizes of the molecules. For a small solute, the mole fractions would appear instead, and this modification is the innovation due to Flory and Huggins.
is the absolute temperature. The volume fraction is analogous to the mole fraction, but is weighted to take account of the relative sizes of the molecules. For a small solute, the mole fractions would appear instead, and this modification is the innovation due to Flory and Huggins.
Derivation
We first calculate the entropy of mixing, the increase in the uncertainty about the locations of the molecules when they are interspersed. In the pure condensed phases — solvent and polymer — everywhere we look we find a molecule.[3] Of course, any notion of "finding" a molecule in a given location is a thought experiment since we can't actually examine spatial locations the size of molecules. The expression for the entropy of mixing of small molecules in terms of mole fractions is no longer reasonable when the solute is a macromolecular chain. We take account of this dissymmetry in molecular sizes by assuming that individual polymer segments and individual solvent molecules occupy sites on a lattice. Each site is occupied by exactly one molecule of the solvent or by one monomer of the polymer chain, so the total number of sites is
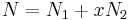
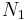 is the number of solvent molecules and
is the number of solvent molecules and 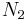 is the number of polymer molecules, each of which has
is the number of polymer molecules, each of which has 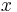 segments.[4]
segments.[4]
From statistical mechanics we can calculate the entropy change, the increase in spatial uncertainty, as a result of mixing solute and solvent.
![\Delta S_m = -k[\,N_1\ln(N_1/N) %2B N_2\ln(xN_2/N)\,]\,](/2012-wikipedia_en_all_nopic_01_2012/I/68d81c2c2e582c5748ebe4d38f8585d8.png)
where 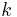 is Boltzmann's constant. Define the lattice volume fractions and
is Boltzmann's constant. Define the lattice volume fractions and
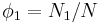
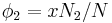
These are also the probabilities that a given lattice site, chosen at random, is occupied by a solvent molecule or a polymer segment, respectively. Thus
![\Delta S_m = -k[\,N_1\ln\phi_1 %2B N_2\ln\phi_2\,]\,](/2012-wikipedia_en_all_nopic_01_2012/I/ad483068b31a8642d6a264ea29b3e725.png)
For a small solute whose molecules occupy just one lattice site, equals one, the volume fractions reduce to molecular or mole fractions, and we recover the usual equation from ideal mixing theory.
In addition to the entropic effect, we can expect an enthalpy change.[5] There are three molecular interactions to consider: solvent-solvent 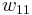 , monomer-monomer
, monomer-monomer 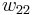 (not the covalent bonding, but between different chain sections), and monomer-solvent
(not the covalent bonding, but between different chain sections), and monomer-solvent 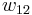 . Each of the last occurs at the expense of the average of the other two, so the energy increment per monomer-solvent contact is
. Each of the last occurs at the expense of the average of the other two, so the energy increment per monomer-solvent contact is
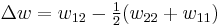
The total number of such contacts is
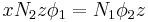
where 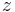 is the coordination number, the number of nearest neighbors for a lattice site, each one occupied either by one chain segment or a solvent molecule. That is,
is the coordination number, the number of nearest neighbors for a lattice site, each one occupied either by one chain segment or a solvent molecule. That is, 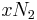 is the total number of polymer segments (monomers) in the solution, so
is the total number of polymer segments (monomers) in the solution, so 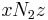 is the number of nearest-neighbor sites to all the polymer segments. Multiplying by the probability that any such site is occupied by a solvent molecule,[6] we obtain the total number of polymer-solvent molecular interactions. An approximation following mean field theory is made by following this procedure, thereby reducing the complex problem of many interactions to a simpler problem of one interaction.
is the number of nearest-neighbor sites to all the polymer segments. Multiplying by the probability that any such site is occupied by a solvent molecule,[6] we obtain the total number of polymer-solvent molecular interactions. An approximation following mean field theory is made by following this procedure, thereby reducing the complex problem of many interactions to a simpler problem of one interaction.
The enthalpy change is equal to the energy change per polymer monomer-solvent interaction multiplied by the number of such interactions
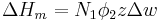
The polymer-solvent interaction parameter chi is defined as
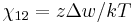
It depends on the nature of both the solvent and the solute, and is the only material-specific parameter in the model. The enthalpy change becomes
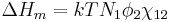
Assembling terms, the total free energy change is
where we have converted the expression from molecules and to moles and by transferring Avogadro's number 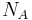 to the gas constant
to the gas constant 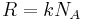 .
.
The value of the interaction parameter can be estimated from the Hildebrand solubility parameters 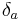 and
and 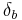
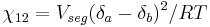
where 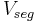 is the actual volume of a polymer segment.
is the actual volume of a polymer segment.
This treatment does not attempt to calculate the conformational entropy of folding for polymer chains. (See the random coil discussion.) The conformations of even amorphous polymers will change when they go into solution, and most thermoplastic polymers also have lamellar crystalline regions which do not persist in solution as the chains separate. These events are accompanied by additional entropy and energy changes.
More advanced models exist, such as the Flory-Krigbaum theory.
External links
- "Conformations, Solutions and Molecular Weight" (book chapter), Chapter 3 of Book Title: Polymer Science and Technology; by Joel R. Fried; 2nd Edition, 2003
- Flory-Huggins in practice: discussion on solubility in polymer based materials
References and footnotes
- ^ "Thermodynamics of High Polymer Solutions," Paul J. Flory Journal of Chemical Physics, August 1941, Volume 9, Issue 8, p. 660 Abstract. Flory suggested that Huggins' name ought to be first since he had published several months earlier: Flory, P.J., "Thermodynamics of high polymer solutions," J. Chem. Phys. 10:51-61 (1942) Citation Classic No. 18, May 6, 1985
- ^ "Solutions of Long Chain Compounds," Maurice L. Huggins Journal of Chemical Physics, May 1941 Volume 9, Issue 5, p. 440 Abstract
- ^ We are ignoring the free volume due to molecular disorder in liquids and amorphous solids as compared to crystals. This, and the assumption that monomers and solute molecules are really the same size, are the main geometric approximations in this model.
- ^ For a real synthetic polymer, there is a statistical distribution of chain lengths, so would be an average.
- ^ The enthalpy is the internal energy corrected for any pressure-volume work at constant (external)
 . We are not making any distinction here. This allows the approximation of Helmholtz free energy, which is the natural form of free energy from the Flory-Huggins lattice theory, to Gibbs free energy.
. We are not making any distinction here. This allows the approximation of Helmholtz free energy, which is the natural form of free energy from the Flory-Huggins lattice theory, to Gibbs free energy. - ^ In fact, two of the sites adjacent to a polymer segment are occupied by other polymer segments since it is part of a chain; and one more, making three, for branching sites, but only one for terminals.
|
||||||||||||||